Running the pipeline
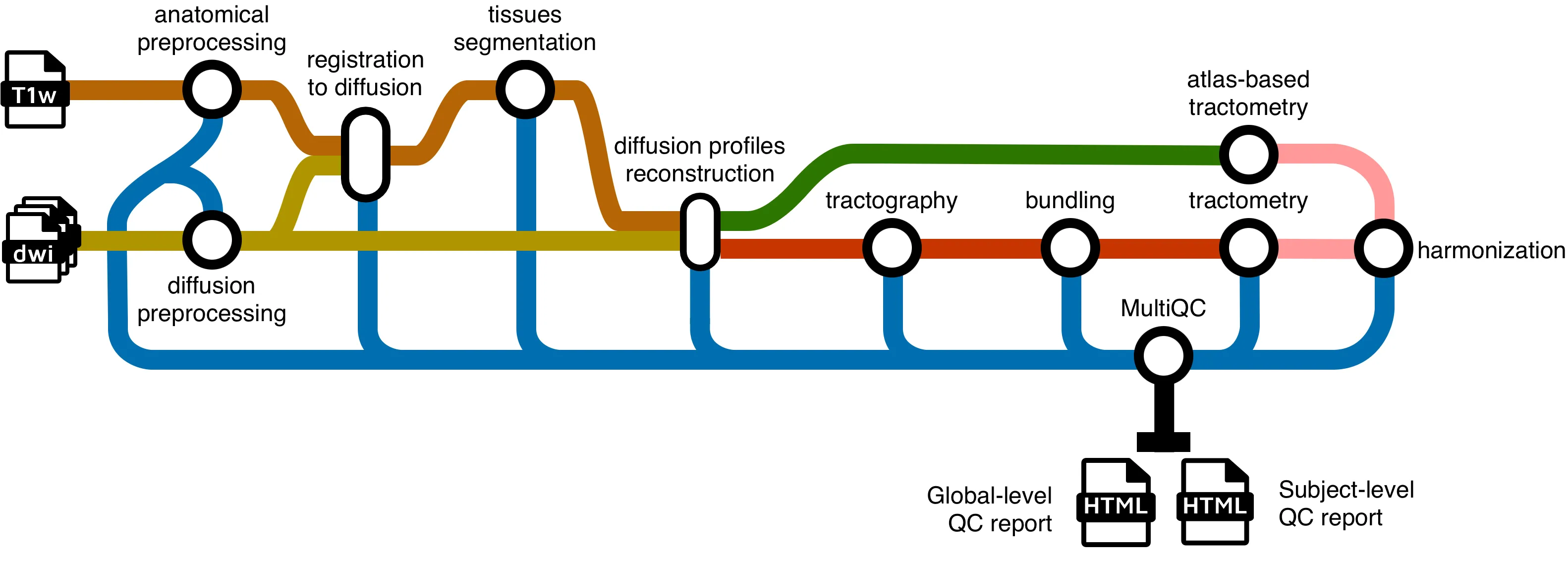
Running the pipeline
Section titled “Running the pipeline”The typical command for running the pipeline locally is as follows:
nextflow run scilus/sf-tractomics -r <release_version> \ --input <input_directory> \ --outdir ./results \ -profile docker,full_pipeline \ -with-report <report_name>.html \ -resumeThis will launch the pipeline with the docker and the full_pipeline configuration profiles, which automatically spawns docker containers when necessary and runs all the possible diffusion MRI processing steps respectively.
-
--input: the path to your BIDS directoryFor more details on how to organize your input folder, please refer to the inputs section.
-
--outdir: path to the output directoryWe do not specify a default for the output directory location to ensure that users have total control on where the output files will be stored, as it can quickly grow into a large number of files. The recommended naming would be something along the line of
sf-tractomics-v{version}where{version}could be0.1.0for example. -
-profile: profile(s) to be run and container system to useThis is a core
nextflowargument.sf-tractomicsprocessing steps was designed in profiles, giving users total control on which type of processing they want to make. One caveat is that users need to explicitly tell which profile to run. This is done via the-profileparameter.Multiple profiles can be specified at once by separating them with only a comma and no whitespace (important!). To choose the appropriate profile(s) for your needs, please see this section.
-
-with-report: Enablesnextflowcaching capabilities.This is a core
nextflowargument. It enables the creation of a html report of the pipeline execution. This report includes some basic metrics about a pipeline run. For more details, see the corenextflowarguments section. -
-resume: Enablesnextflowcaching capabilities.This is a core
nextflowargument. It enables the resumability of your pipeline. In the event where the pipeline fails for a variety of reasons, the following run will start back where it left off. For more details, see the corenextflowarguments section.
Note that the pipeline will create the following files in your working directory:
Directorywork/ # Nextflow working directory
- …
- .nextflow_log # Log file from Nextflow
Directorysf-tractomics-v0.1.0/ # Results location (defined with —outdir)
- pipeline_info # Global informations on the run
- stats # Global statistics on the run
Directorysub-01
… # Other entities like session
Directoryanat/ # Clean T1w in diffusion space
- …
Directorydwi/ # All clean DWI files, models, tractograms, …
Directorybundles/ # Extracted bundles when using bundling
- …
Directoryxfm/ # Transforms between diffusion and anatomy
- …
- … # Other nextflow related files
Choosing a profile
Section titled “Choosing a profile”Some sf-tractomics core functionalities are accessed and selected using profiles and arguments.
Configuration profiles:
-
-profile docker(Recommended):Each process will be run using Docker containers.
-
-profile apptainer(Recommended):Each process will be run using Apptainer images.
-
-profile slurm:If selected, the SLURM job scheduler will be used to dispatch jobs. This is most commonly used when running pipelines on an HPC server.
-
-profile arm:Made to be use on computers with an ARM architecture (e.g., Mac M-series chips). This is still experimental and some containers might not be built for the ARM architecture yet. Feel free to open an issue if needed.
Processing profiles:
-
-profile full_pipelineRuns the full sf-tractomics pipeline from end-to-end, with all processing steps enabled.
-
-profile iit_tractometryPreprocesses the inputs (denoising, registration, etc.), computes diffusion metric maps, freewater correction and NODDI for multi-shell subjects. This profile performs tractometry without tracking. It registers the bundle definitions (i.e. TDI maps or binary masks) from the IIT White Matter Atlas to the subject space to perform tractometry.
-
-profile gpuActivate usage of GPU accelerated algorithms to drastically increase processing speeds. Currently only supports CUDA (e.g. NVidia’s GPU). Accelerations for FSL Eddy and Local tractography are automatically enabled using this profile. If you’re encountering errors while using this profile, please refer to the troubleshooting page.
Using either -profile docker or -profile apptainer is highly recommended, as it controls the version of the software used and ensure reproducibility. While it is technically possible to run the pipeline without Docker or Apptainer, the amount of dependencies to install is simply not worth it.
Key parameters
Section titled “Key parameters”Shell selection for DTI and fODF models
Section titled “Shell selection for DTI and fODF models”By default, the pipeline assumes that:
-
B-values ≤ 1200 s/mm² are used for DTI fitting.
-
B-values ≥ 700 s/mm² are used for fODF fitting.
If your data has different b-values, you MUST specify which shells to use, or the pipeline will likely fail. To do that, you can change the thresholds used for the DTI and fODF fitting by specifying the following parameters: --max_dti_shell_value and --min_fodf_shell_value depending on your use case. For example, if your data has a protocol where the b-values under 1500 s/mm² should be used for DTI fitting (e.g. your protocol doesn’t have b-values under 1200 s/mm²) and the b-values above 800 s/mm² should be used for fODF fitting, running the pipeline would look something like:
nextflow run scilus/sf-tractomics -r <release_version> \ --input <input_directory> \ --outdir ./results \ --max_dti_shell_value 1500 \ --min_fodf_shell_value 800 \ [...]For more details, see the parameters page.
Include/exclude subjects
Section titled “Include/exclude subjects”The pipeline will try to process all the subjects it can find within your input BIDS folder. However, for many different reasons, you might not want to process all of your subjects.
-
Include subjects: You might want to run the pipeline on a subset of your subjects (e.g. to test, or to process your data in many batches). You can specify the
--participant_labelparameter with a list of comma-separated subject labels. This will only run the pipeline on those subjects. For example, to only include the subjects “sub-02” and “sub-03”, running the pipeline would look something like:Terminal window nextflow run scilus/sf-tractomics -r <release_version> \--input <input_directory> \--outdir ./results \--participant_label "sub-02,sub-03"[...] -
Exclude subjects: If you already know that you will exclude subjects from your analysis, you can specify the
--exclude_participant_labelparameter with a list of comma-separated subject labels. This will avoid running the pipeline on those subjects. For example, to exclude the subjects “sub-01”, “sub-04” and “sub-05”, the command would look something like:Terminal window nextflow run scilus/sf-tractomics -r <release_version> \--input <input_directory> \--outdir ./results \--exclude_participant_label "sub-01,sub-04,sub-05"[...]
You can also specify both parameters at the same time. The inclusion of the subjects will be made first, followed by the exclusion. If a specified subject doesn’t exist in the input dataset, it will simply be skipped.
Core nextflow arguments
Section titled “Core nextflow arguments”-profile
Section titled “-profile”Use this parameter to choose a configuration profile. Profiles can give configuration presets for different compute environments.
Several generic profiles are bundled with the pipeline which instruct the pipeline to use software packaged using different methods (Docker, Singularity, and Apptainer) - see below.
The pipeline also dynamically loads configurations from https://github.com/nf-core/configs when it runs, making multiple config profiles for various institutional clusters available at run time. For more information and to check if your system is supported, please see the nf-core/configs documentation.
Note that multiple profiles can be loaded, for example: -profile tracking,docker - the order of arguments is important!
They are loaded in sequence, so later profiles can overwrite earlier profiles. For a complete description of the available profiles, please see this
section.
-resume
Section titled “-resume”Specify this when restarting a pipeline. Nextflow will use cached results from any pipeline steps where the inputs are the same, continuing from where it got to previously. For input to be considered the same, not only the names must be identical but the files’ contents as well. For more info about this parameter, see this blog post.
You can also supply a run name to resume a specific run: -resume [run-name]. Use the nextflow log command to show previous run names.
-with-report
Section titled “-with-report”Nextflow can create an HTML execution report: a single document which includes many useful metrics about a workflow execution. The report is organised in the three main sections: Summary, Resources and Tasks.
-params-file
Section titled “-params-file”Instead of specifying all your pipeline parameters one by one each time in the command line when running the pipeline, you can specify your parameters in a single configuration file. Note that this doesn’t work for nextflow core arguments (with single hyphens). The parameters file can either be a YAML or JSON. For example:
input: '<input_directory>/'outdir: './results/'max_dti_shell_value: 1500min_fodf_shell_value: 800participant_label: "sub-02,sub-03"<...>And running the pipeline with:
nextflow run scilus/sf-tractomics -r <release_version> \ -params-file params.yaml \ [...]{ "input": "<input_directory>/", "outdir": "./results/", "max_dti_shell_value": 1500, "min_fodf_shell_value": 800, "participant_label": "sub-02,sub-03"}And running the pipeline with:
nextflow run scilus/sf-tractomics -r <release_version> \ -params-file params.json \ [...]This specifies a path to a nextflow configuration file. This file differs from the previous -params-file argument since this allows to tune process resource specifications, other infrastructural tweaks (such as output directories and published files) or module arguments (args). Beware that this way of customizing the pipeline’s execution might require a more in-depth knowledge of the pipeline and it is very error-prone. The following example simply illustrates how to structure such a configuration:
process { withName: ".*:ENSEMBLE_TRACKING" { memory = 16.GB ext.suffix = "ensemble_tracking" publishDir = false }}And running the pipeline with:
nextflow run scilus/sf-tractomics -r <release_version> \ -c nextflow.config \ [...]Troubleshooting
If you encountered any issues when following the steps detailed in this page, refer to the troubleshooting page or open an issue if your error is not covered (yet).